1. 订书肽发展简介
订书肽是基于多肽需形成α-螺旋通过细胞膜进入细胞的需求上发展起来的。生物体内的多种生命进程调节都是通过蛋白质与蛋白质之间的相互作用来实现的。例如病毒的自组装,细胞的生长,分裂,分化等过程。而通常蛋白-蛋白相互作用的界面太大,从而使小分子药物很难对其进行靶向定位,达到高效特异性地阻断这种相互作用,展现良好的治疗效果。蛋白类药物因为很难顺利通过细胞膜所以也达不到直接靶向细胞内相互作用的效果,因此,研究者们开始寻求一种能够克服这两种药物缺点的既能够进入细胞膜又能特异性靶向蛋白-蛋白相互作用的新的药物分子。
研究表明,具有α-螺旋结构和富含正电荷的多肽可以穿过细胞膜。因此,人们开发了利用二硫键与分子内酰胺键作为支架的α-螺旋结构,但是,这些支架在生理环境下均不能稳定存在。2000年,Verdine等发展了一种用碳碳键作为支架来稳定多肽α-螺旋结构的方法,由此方法得到的多肽成为订书肽(Stapled
peptides)。订书肽有更高的α-螺旋程度,结合能力强,能通过细胞膜,难被蛋白酶水解,在生物体内半衰期长等优点。
2. 订书肽合成
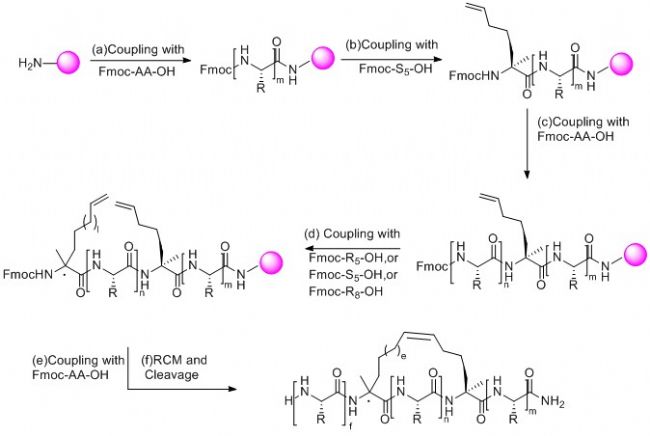
订书肽合成策略为在固相合成肽链过程中引入两个含有α-甲基,α-烯基的非天然氨基酸,然后两个非天然氨基酸之间发生烯烃复分解反应(RCM)环化构成稳定α-螺旋结构构象的全碳支架,从而合成订书肽。
1) α-甲基,α-烯基的非天然氨基酸的合成:

α-甲基,α-烯基的非天然氨基酸的一般结构
通常,在三乙胺的催化下,手性辅基试剂(1R,2S)-2-氨基-1,2-二苯乙醇与2-溴丙酸乙酯发生亲核取代反应,并采用(Boc)2O保护自由胺基;接下来,在对甲苯磺酸的催化下,环状的含有手性辅基的丙氨酸前体得以高效生产,在碱性催化剂和低温下,烯基基团高效立体选择性的连接到氨基酸的α-位,最后,在氨基锂条件下脱去手性辅基,并采用Fmoc保护α-胺基。

α-甲基,α-烯基的非天然氨基酸的合成路线
2) 订书肽的合成:
通常采用Fmoc-固相多肽合成方法制备订书肽,两个用于构象锁定的α-甲基,α-烯基的非天然氨基酸之间一般间隔两个,三个或者六个氨基酸,多肽合成完毕后,采用钌作催化剂进行烯烃复分解反应(RCM),如果实验需要,还可以进行生物素(Biotin),荧光染色等修饰,最后,将目标多肽从树脂上切割下来进行纯化得到成品。